160 TMA-4¶
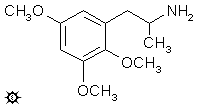
2,3,5-三甲氧基苯丙胺 (2,3,5-TRIMETHOXYAMPHETAMINE)
 |
|---|
合成: 将 68 g 2,4-二甲氧基苯甲醛溶解在 250 mL 冰乙酸中,加热至 25 °C 并充分搅拌,向其中逐滴加入 86 g 40% 的过氧乙酸溶液(溶于乙酸)。该反应是放热的,滴加速度需控制以保持内部温度在 28 °C 左右几度的范围内。根据需要使用外部冷却。滴加耗时 1 小时,当反应明显完成后(温度不再上升),将整个反应混合物倒入 3 倍体积的水中。过量的酸用固体碳酸钾中和(需要 283 g)。用 3x100 mL 乙醚提取,合并提取液,并在真空下除去溶剂,得到 66 g 粗制甲酸 2,4-二甲氧基苯酯。将其悬浮在 125 mL 10% 氢氧化钠中,混合物在蒸汽浴上加热 1.5 小时。冷却后,反应混合物凝固成厚重的黑色固体。过滤除去固体,用水洗涤,并溶解在 250 mL 二氯甲烷中。有机相用稀盐酸洗涤,然后用碳酸氢钠水溶液洗涤,这除去了大部分颜色。在真空下除去溶剂得到深红色粘稠物,将其溶解在 200 mL 无水乙醚中并通过滤纸过滤。除去所得澄清溶液的溶剂,得到 34.4 g 2,4-二甲氧基苯酚,为红色油状物,冷却后结晶。取 1.0 g 样品溶于 4 mL 吡啶中,用 0.9 g 苯甲酰氯处理并在蒸汽浴上加热几分钟。加入水得到糊状固体,通过在多孔板上压制分离。粗制苯甲酸 2,4-二甲氧基苯酯的产量为 1.1 g。从环己烷中重结晶得到白色产物,熔点为 86-87 °C。从环己烷中第二次重结晶将熔点提高到 89-90 °C,这与文献值一致。
向 31.0 g 粗制 2,4-二甲氧基苯酚在 60 mL 无水乙醇的溶液中,加入 11.25 g 氢氧化钾在 90 mL 沸腾乙醇中的溶液。然后向其中加入 28 g 烯丙基溴,立即产生白色溴化钾沉淀。混合物回流 2 小时,然后倒入 3 倍体积的水中。加入足量的 10% 氢氧化钠使反应液呈强碱性,用 3x100 mL 乙醚提取。在真空下除去溶剂得到 33.2 g 1-烯丙氧基-2,4-二甲氧基苯,气相色谱分析显示不含苯酚原料。分析必须在低柱温(低于 180 °C)下于乙二醇琥珀酸酯基质上进行。如果使用硅酮柱,即使在这些低温下,柱上也会发生相当大的克莱森重排。可以使用低温蒸馏进行进一步纯化(1.0 mm/Hg 下 107-110 °C)。
将 31.0 g 1-烯丙氧基-2,4-二甲氧基苯样品用微火缓慢加热,直到内部温度达到 215 °C。发生放热反应,温度升至 270 °C。烧瓶中残留的主要是 2-烯丙基-4,6-二甲氧基苯酚,其中可能含有 10% 的 2,4-二甲氧基苯酚,这是由于热解失去烯丙基基团所致。该混合物未经进一步纯化即进行甲基化。
向 30 g 不纯的 2-烯丙基-4,6-二甲氧基苯酚在少量无水乙醇的溶液中,加入 8.7 g 氢氧化钾在 75 mL 无水乙醇中的沸腾溶液,紧接着加入 22.4 g 碘甲烷在少量乙醇中的溶液。混合物回流 3 小时,然后加入 4 倍体积的水中。加入足量的 10% 氢氧化钠使混合物呈强碱性,用 4x100 mL 乙醚提取。除去溶剂得到 28 g 1-烯丙基-2,3,5-三甲氧基苯。气相色谱分析显示含有约 10% 的预期杂质 1,2,4-三甲氧基苯。
向 26 g 粗制 1-烯丙基-2,3,5-三甲氧基苯在等重量无水乙醇的溶液中,加入 52 g 片状氢氧化钾。混合物在蒸汽浴上加热过夜,然后用大量水稀释。用 3x100 mL 乙醚提取,真空除去溶剂后得到 24.6 g 产物。通过气相色谱分析,主要包含顺式和反式-1-丙烯基-2,3,5-三甲氧基苯以及预期的 1,2,4-三甲氧基苯。将该混合物溶解在等体积的戊烷中,并在干冰中冷却。快速过滤得到 9.2 g 琥珀色固体,熔点为 39-41.5 °C。从己烷中重结晶得到纯的反式-1-丙烯基-2,3,5-三甲氧基苯,熔点为 44-45 °C。蒸发原始的戊烷母液得到不纯的顺式和反式异构体混合样品。
将 7.2 g 反式-1-丙烯基-2,3,5-三甲氧基苯溶于 41 g 干丙酮的溶液用 3.3 g 干吡啶处理,并在充分搅拌下冷却至 0 °C。然后在 1 分钟内加入 6.9 g 四硝基甲烷,反应混合物再搅拌 2 分钟。然后用 2.2 g 氢氧化钾溶于 40 mL 水的溶液淬灭反应混合物。加入更多水后,用 3x50 mL 二氯甲烷提取产物。在真空下除去溶剂得到 7.0 g 不纯产物,无法结晶。将其在真空下蒸馏得到四个馏分,所有馏分均自发结晶。馏分 #1 和 #2(2 mm/Hg 下沸点 100-120 °C 和 120-130 °C)合并,重 0.8 g,从己烷中结晶后得到白色晶体,熔点为 62-63 °C。NMR 谱(在 CDCl3 中)与 2,3,5-三甲氧基苯甲醛一致,文献报道的熔点为 62-63 °C。馏分 #3 和 #4(2 mm/Hg 下沸点 130-170 °C 和 170-175 °C,大部分在后一馏分中馏出)合并得到 3.0 g 黄色晶体。这些晶体在少量冷甲醇下研磨,然后从甲醇中重结晶,得到 1.15 g 2-硝基-1-(2,3,5-三甲氧基苯基)丙烯黄色晶体,熔点为 87-88 °C。气相色谱分析显示,蒸馏的前馏分含有相当多未反应的反式-1-丙烯基-2,3,5-三甲氧基苯和一些 1,2,4-三甲氧基苯。
向回流并搅拌的 1.1 g 氢化铝锂在 150 mL 无水乙醚的悬浮液中(惰性气氛下),加入 1.1 g 2-硝基-1-(2,3,5-三甲氧基苯基)丙烯在 50 mL 无水乙醚中的溶液。乳状混合物回流 4 小时,冷却,然后通过加入 1.5 N 硫酸小心地破坏过量的氢化物。然后加入 20 g 酒石酸钾钠,接着加入足量的氢氧化钠水溶液使 pH >9。分离乙醚相,剩余的水相用 3x75 mL 二氯甲烷提取。合并有机相和提取液,真空除去溶剂,得到 0.9 g 无色油状物。将其溶解在 200 mL 饱和了无水氯化氢气体的无水乙醚中。生成了不结晶的浓稠油状物。倒出乙醚,让油状物在室温下的密封容器中放置几天。析出了细小的白色针状 2,3,5-三甲氧基苯丙胺盐酸盐 (TMA-4),经乙醚洗涤和风干后重 0.31 g。熔点为 118-119 °C。分析 (C12H20ClNO3) C,H。残留的油状物溶于水,用氢氧化钠碱化,并用二氯甲烷提取。蒸发溶剂得到 0.40 g 白色油状物,将其溶解在含有 0.22 g 草酸的少量甲醇中。立即析出 2,3,5-三甲氧基苯丙胺草酸盐晶体,熔点约为 110 °C。
给药剂量: 大于 80 mg。
药效时长: 也许 6 小时。
定性评论: (80 mg)我关注生活问题,进行了大量的内省,持续约 6 小时。没有主观的身体症状。对我来说,它相当于约 50 微克的 LSD,或 120 毫克的 TMA。
延伸和评论: 这就是现存关于主观效应的全部知识。最终得到的盐酸盐极其珍贵且少量,当完成所需的剂量累积时,剩下的仅够在南美进行的这一次试验。基于自愿者将其与 LSD 和 TMA 的比较,已发表的关于该化合物效力的说法称其为麦司卡林效力的 4 倍,即 4 M.U.。该物质必须重新合成,并按照现在接受的协议重新评估。
在未来的重新合成中,上述几个步骤将有相当大的改进。苯酚的制备、烯丙基醚、克莱森重排、新苯酚的甲基化以及异构化为顺式和反式丙烯基苯混合物的产物,都是在没有 Kugel-Rohr 装置的情况下进行的。产物变得越来越稠和黑,幸亏在反式丙烯基阶段得到了固体,才最终获得了一定程度的纯度。所有的中间体肯定都是白色油状物,当重复此制备时,它们将在每一个阶段进行蒸馏。
芳环上甲氧基基团的这种 2,3,5-取向是化学家已知的三取代模式中最困难的一种。根本没有任何简单的方法将其组合在一起。2-碳苯乙胺(2,3,5-三甲氧基苯乙胺)很久以前就已合成。它作为体外研究中肝胺氧化酶底物的作用已被探索,但从未在人体中试验过。更奇怪的是具有这种氧化模式的苯丙胺,其中亚甲二氧基环取代了两个相邻的甲氧基。这种物质是 2,3-亚甲二氧基-5-甲氧基苯丙胺,或 MMDA-4。尽管它具有理论上的吸引力(作为六种可能的 MMDA 衍生物之一)和合成挑战(如上面的 2,3,5-三甲氧基化合物一样,所有东西的位置都不对),但该化合物的药理学未知。这很合逻辑,因为它从未被合成过。没人能设计出可行的程序来制造它。在将 MMDA 的所有可能位置异构体明确列为附表 I 药物的过程中,DEA 命名了这种化合物,既然它被特别命名,就被输入了化学文摘。所以它在文献中有列出,至少在化学文摘中是这样。但实际上它完全不为人知。总有一天,某处的某人会灵光一闪,找到一种合成工艺来制造它。当然,一旦它被制造出来,只要目前的法律保持不变,这就是一种非法行为,至少在美国是这样,因为它目前是附表 I 药物。
不用说,MMDA-4 的 2-碳类似物,2,3-亚甲二氧基-5-甲氧基苯乙胺(2C-MMDA-4 会是一个合理的名称吗?)也是未知的。