拮抗剂¶
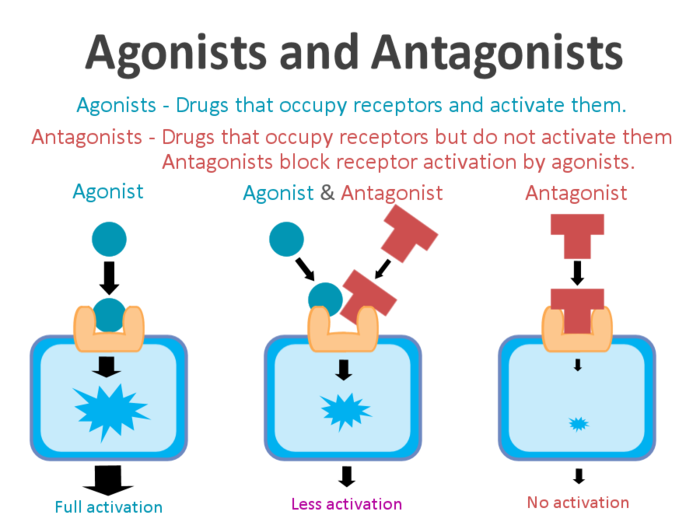
上图展示了激动剂和拮抗剂之间的区别。
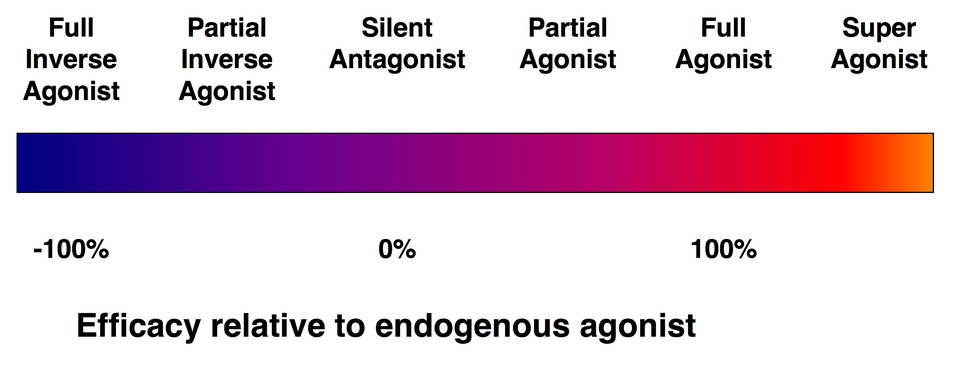
受体配体的效力谱
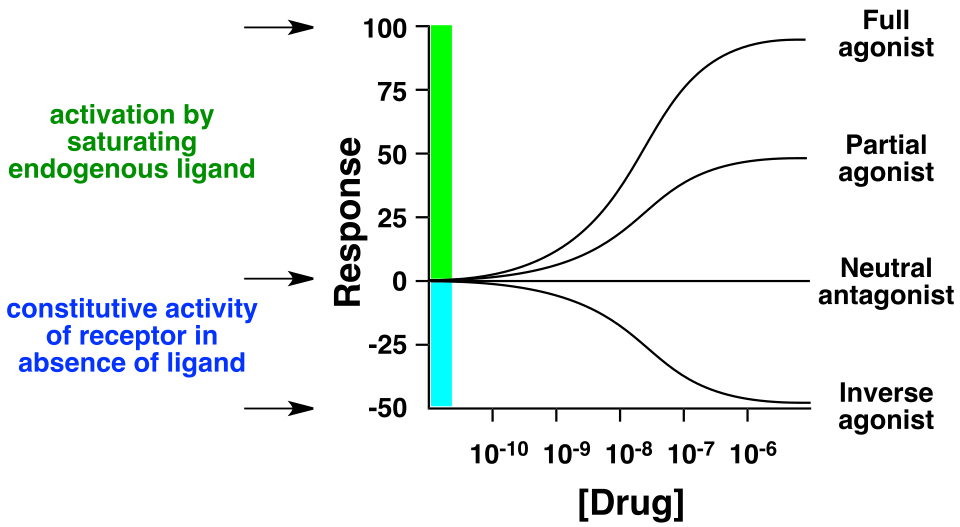
受体配体的剂量反应曲线
**受体拮抗剂**是一种结合细胞受体但不会触发该细胞反应的化学物质。拮抗剂对它们所靶向的受体具有亲和力,但没有激活受体的效力。[1][2] 在没有受体激动剂的情况下,受体拮抗剂没有活性;然而,受体拮抗剂的结合会破坏相互作用并抑制激动剂或逆向激动剂在受体上的功能。
拮抗剂通过结合它们所靶向的受体上的不同位置来介导其作用。一些拮抗剂靶向与受体激动剂相同的结合位点,而另一些则结合受体上的变构位点;拮抗剂也可能在通常不参与受体活性调节的独特结合位点相互作用。受体拮抗剂与其靶向受体的结合可能是永久的或暂时的,这取决于所形成键的性质。
根据它们与受体结合的位置和性质,拮抗剂可细分为竞争性或非竞争性,以及可逆或不可逆。
目录¶
拮抗剂的类型¶
竞争性拮抗剂¶
**竞争性拮抗剂**可逆地结合到与内源性配体或激动剂相同的受体结合位点,而不激活受体。竞争性拮抗剂与激动剂竞争受体上的相同结合位点。当结合到受体上时,竞争性拮抗剂将阻断激动剂的结合。
在激动剂和竞争性拮抗剂同时存在的情况下,靶受体的活性程度取决于每种药物对活性结合位点的相对亲和力及其各自的浓度。在控制浓度的情况下,亲和力较高的药物将比亲和力较低的药物占据更多的受体。通过充分增加受体激动剂的浓度,可以降低或消除竞争性拮抗剂阻断激动剂作用的能力。[3] 因此,竞争性拮抗剂不会降低受体激动剂的最大反应,但确实会增加达到该反应所需的激动剂剂量。[4]
纳洛酮是靶向阿片受体的竞争性受体拮抗剂的一个例子。纳洛酮通过对阿片受体具有比阿片类激动剂高得多的亲和力来实现其效果,从而在受体占有率上胜过激动剂。[5][6]
不可逆竞争性拮抗剂¶
**不可逆竞争性拮抗剂**靶向受体的活性位点,最初与受体激动剂竞争结合受体。然而,一旦结合,不可逆竞争性拮抗剂就会永久地使受体失活。[7] 失活的受体不能受到任何数量的受体激动剂的影响,因此降低了激动剂对一组受体可能产生的最大影响,直到身体合成新的受体。不可逆拮抗剂的持续时间取决于靶受体组的合成速率,该速率因受体类型而异。不可逆竞争性拮抗剂有时因其降低激动剂最大可实现效果的能力而被归类为非竞争性拮抗剂。[7]
在某些科的毒蛇中发现的 α-神经毒素是不可逆竞争性拮抗作用的一个例子。α-神经毒素竞争性且不可逆地结合神经肌肉突触中的烟碱型乙酰胆碱受体(nAChRs)。[8] 不可逆竞争性拮抗剂也用于医学,例如纳洛肼(不要与纳洛酮混淆)。纳洛肼通过共价键不可逆地结合 μ-阿片受体,使分子无法解离并永久阻断受体。
非竞争性拮抗剂¶
**非竞争性拮抗剂**结合它所靶向受体的变构位点,或者是激动剂结合的活性位点以外的位点。这种类型的结合是非竞争性的,因为受体拮抗剂不必与激动剂竞争结合。非竞争性拮抗剂通过诱导受体的构象变化产生其效果,这种变化改变了活性结合位点的形状,阻止激动剂结合。无论存在多少激动剂,非竞争性拮抗作用都无法被抵消,因此在拮抗剂保持与受体结合期间,会降低受体激动剂的最大可能效果。非竞争性拮抗剂通常是可逆的,但也可能是不可逆的。
部分激动剂¶
部分激动剂在全激动剂存在的情况下可能充当竞争性拮抗剂。部分激动剂对其靶向受体的效力低于全激动剂,因此产生较弱的效果。在部分激动剂存在的情况下,全激动剂必须竞争受体占有率。这导致一些受体被部分激动剂占据,一些被全激动剂占据,产生的总体反应比单独使用全激动剂产生的反应要弱。部分激动剂的效果可以通过增加全激动剂的剂量来克服。
丁丙诺啡是 μ-阿片受体的部分激动剂,常用于较高剂量下治疗阿片类药物成瘾。单独使用时,它是 μ-阿片受体的弱激动剂,具有高亲和力和低效力;然而,在其他阿片类药物存在的情况下,丁丙诺啡具有拮抗作用。足够剂量的丁丙诺啡可以通过阻止它们结合 μ-阿片受体,来减弱或抵消甚至大剂量的可待因、吗啡或海洛因的额外效果。[10] 在服用丁丙诺啡的患者中很难实现药物性阿片镇痛,需要一种具有更高亲和力的药物来胜过它以结合 μ-阿片受体,例如芬太尼或其类似物。[11]
可逆性¶
受体拮抗剂可根据其与靶向受体结合的性质和持续时间分类为可逆或不可逆。可逆拮抗剂像大多数激动剂一样,随着时间的推移容易与受体结合和解离。不可逆拮抗剂与其靶向的受体形成稳定的永久键,并且无法解离。受体和拮抗剂复合物结合的倾向由解离常数描述。不可逆拮抗剂由于其极高的亲和力,解离常数为 0(或实际上如此)。可逆拮抗剂与其受体形成较弱的键,因此它们具有较低的亲和力和较高的解离率。[2]
另见¶
外部链接¶
参考文献¶
- ↑ Stephenson, R. P. (February 1997). "A MODIFICATION OF RECEPTOR THEORY". British Journal of Pharmacology. 120 (Suppl 1): 106–120. doi:10.1111/j.1476-5381.1997.tb06784.x. ISSN 0007-1188.
- ↑ 2.0 2.1 Drug–Receptor Interactions - Clinical Pharmacology
- ↑ Swinney, D. C. (September 2004). "Biochemical mechanisms of drug action: what does it take for success?". Nature Reviews Drug Discovery. 3 (9): 801–808. doi:10.1038/nrd1500. ISSN 1474-1784.
- ↑ Dose response curves in the presence of antagonists | http://www.curvefit.com/schild.htm
- ↑ Naloxone Hydrochloride - The American Society of Health-System Pharmacists | http://www.drugs.com/monograph/naloxone-hydrochloride.html
- ↑ Sirohi, S., Dighe, S. V., Madia, P. A., Yoburn, B. C. (August 2009). "The relative potency of inverse opioid agonists and a neutral opioid antagonist in precipitated withdrawal and antagonism of analgesia and toxicity". The Journal of Pharmacology and Experimental Therapeutics. 330 (2): 513–519. doi:10.1124/jpet.109.152678. ISSN 1521-0103.
- ↑ 7.0 7.1 http://www.merckvetmanual.com/mvm/pharmacology/pharmacology_introduction/drug_action_and_pharmacodynamics.html#v3329185
- ↑ Young, H. S., Herbette, L. G., Skita, V. (August 2003). "α-Bungarotoxin Binding to Acetylcholine Receptor Membranes Studied by Low Angle X-Ray Diffraction". Biophysical Journal. 85 (2): 943–953. ISSN 0006-3495.
- ↑ Kohrs, R., Durieux, M. E. (November 1998). "Ketamine: Teaching an Old Drug New Tricks". Anesthesia & Analgesia. 87 (5): 1186–1193. doi:10.1213/00000539-199811000-00039. ISSN 0003-2999.
- ↑ Alford, D. P., Compton, P., Samet, J. H. (17 January 2006). "Acute Pain Management for Patients Receiving Maintenance Methadone or Buprenorphine Therapy". Annals of internal medicine. 144 (2): 127–134. ISSN 0003-4819.
- ↑ Rosenquist, R. W., Souzdalnitski, D., Urman, R. D. (2016). Chronic Pain Management for the Hospitalized Patient. Oxford University Press. ISBN 9780199349302.