75 EME¶
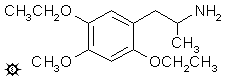
2,5-二乙氧基-4-甲氧基苯丙胺
 |
|---|
合成: 向 14.0 g 4-乙氧基-3-甲氧基苯酚(该起始原料的制备见 MME 的配方)溶于等体积乙醇的溶液中,加入 5.3 g 氢氧化钾溶于 100 mL 热甲醇的溶液。随后加入 9.1 g 溴乙烷,混合物保持回流 2 小时。5 分钟内即有明显的溴化钾沉积,反应结束时出现相当严重的暴沸。混合物用 3 倍体积的水和 1 倍体积的 5% 氢氧化钠稀释,并用 2x200 mL 乙醚提取。合并提取液,真空除去溶剂,得到 14.3 g 淡琥珀色油状物,固化为 2,5-二乙氧基苯甲醚晶体,熔点为 44-45 °C。文献报道该化合物由硫酸二乙酯作用于甲氧基对苯二酚制得。
将 24.1 g N-甲基甲酰苯胺和 27.3 g 三氯氧磷的混合物在室温下静置直至呈现强烈的红色(约 0.5 小时),然后加入 13.8 g 固体 2,5-二乙氧基苯甲醚,混合物在蒸汽浴上加热 2 小时。将黑色、粘稠的反应产物倒在碎冰上,并不停搅拌,颜色变浅,形成淡黄色粉末。静置几小时后,过滤除去并尽可能吸干。32 g 潮湿产物通过气相色谱显示存在同分异构体污染,水母液经二氯甲烷提取和浓缩后,显示出更多类似醛的杂质。分离出的固体从 125 mL 沸腾的甲醇中重结晶,得到 15.8 g 淡黄色晶体(湿重),气相色谱仍显示可检测到的杂质。从 100 mL 沸腾的甲醇中进行第二次重结晶,得到灰白色蓬松晶体 2,5-二乙氧基-4-甲氧基苯甲醛,风干后重 8.5 g。熔点为 109-110 °C。两次甲醇结晶的合并母液除去溶剂,所得固体块再次从甲醇中结晶,得到第二批醛,5.7 g,熔点为 110-111 °C。将 1.0 g 该醛和 0.7 g 丙二腈溶于 40 mL 温无水乙醇的溶液用几滴三乙胺处理。大约一分钟后,形成晶体。过滤除去这些晶体,用乙醇洗涤,并风干,得到 0.6 g 2,5-二乙氧基-4-甲氧基苯亚甲基丙二腈,为明亮的黄色晶体,熔点为 156.5-158 °C。
将 6.7 g 2,5-二乙氧基-4-甲氧基苯甲醛溶于 21 g 冰乙酸的溶液,用 3.1 g 硝基乙烷和 1.93 g 无水乙酸铵处理,并在蒸汽浴上加热 2.5 小时。向热反应混合物中加入少量水,引发橙色产物结晶,混合物恢复至室温并静置数小时后,过滤除去,用水洗涤,并风干。产物 1-(2,5-二乙氧基-4-甲氧基苯基)-2-硝基丙烯呈暗橙色,重 3.0 g,熔点为 84-86 °C。来自甲苯的分析样品熔点为 85-86 °C。元素分析 (C14H19NO5) C,H。
向在氦气气氛下温和回流的 2.0 g 氢化铝锂在 250 mL 无水乙醚中的悬浮液中,加入 2.8 g 1-(2,5-二乙氧基-4-甲氧基苯基)-2-硝基丙烯,方法是让冷凝的乙醚滴入装有硝基苯乙烯的分流索氏提取器套管中。这有效地逐滴加入温热的硝基苯乙烯饱和溶液。添加耗时 1 小时,回流继续进行 6 小时。将反应混合物降至冰浴温度,并通过小心加入 150 mL 1.5 N 硫酸破坏过量的氢化物。当水层和乙醚层最终澄清时,将它们分离,并在水层中溶解 50 g 酒石酸钾钠。然后加入氢氧化钠水溶液直至 pH >9,然后用 3x150 mL 二氯甲烷提取。真空除去溶剂产生 2.3 g 清澈的白色油状物,将其溶于 300 mL 无水乙醚中并通入无水氯化氢气体至饱和。起初溶液保持完全清澈,最后开始形成细小的白色晶体。结晶完成后,过滤除去这些固体,用乙醚洗涤并风干。由此得到 2.2 g 2,5-二乙氧基-4-甲氧基苯丙胺盐酸盐 (EME),熔点为 162-164 °C,先前在 154 °C 软化。元素分析 (C14H24ClNO3) C,H,N。
给药剂量: 未知。
药效时长: 未知。
延伸和评论: 这是 TMA-2 所有可能的乙氧基同系物集合中的另一个。该系列的后部和较重成员是在生物活性方向从早期成员中变得明显之前合成和完成的。这种化合物从未经过测定,合理的猜测是它的效力非常低,在较高剂量水平下有毒性迹象。我怀疑它永远不会被测定,肯定不会由我来测定。