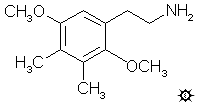
27 2C-G¶
2,5-二甲氧基-3,4-二甲基苯乙胺
 |
|---|
合成: 向400 mL温乙醇中加入40.4 g片状氢氧化钾(KOH),搅拌至澄清,然后加入86.5 g 2,3-二甲苯酚,接着加入51.4 g碘甲烷。该混合物回流2天,真空除去挥发物,残留物溶于1 L水(H2O)中,并用4x200 mL二氯甲烷(CH2Cl2)萃取。合并的萃取液用5%氢氧化钠(NaOH)洗涤,直到洗涤液保持碱性。经过一次稀盐酸(HCl)洗涤后,真空除去溶剂,残留物为41.5 g具有刺激性气味的琥珀色油状物,自发结晶。2,3-二甲基苯甲醚的熔点为25-26 °C,无需进一步纯化即可用于下一步。从碱性水洗液中,经酸化、萃取和除去溶剂后,得到46.5 g粗制的未反应二甲苯酚,可回收利用。
将205 g三氯氧磷(POCl3)和228 g N-甲基甲酰苯胺的混合物置于室温下孵育,直到出现深紫红色并伴有一些自发发热。向其中加入70.8 g 2,3-二甲基苯甲醚,并在蒸汽浴上加热深色反应混合物2.5小时。然后将产物倒入1.7 L水中,搅拌直至自发结晶。过滤除去这些固体,水洗并风干,得到77.7 g棕色晶体状的粗制苯甲醛。将其在0.4 mm/Hg下于70-90 °C蒸馏,得到64.8 g 2,3-二甲基-4-甲氧基苯甲醛,为白色结晶产物,熔点为51-52 °C。用甲醇(MeOH)重结晶得到分析样品,熔点为55-55.5 °C。分析元素(C10H12O2):C,H。丙二腈衍生物(从该醛和丙二腈在乙醇(EtOH)中加一滴三乙胺制得)经乙醇重结晶后熔点为133-133.5 °C。分析元素(C13H12N2O):C,H,N。最近,这种醛已可商业购买。
将32.4 g 2,3-二甲基-4-甲氧基苯甲醛溶于800 mL二氯甲烷中,用58.6 g 85%间氯过氧苯甲酸处理,并回流3天。冷却至室温后,过滤除去白色固体(间氯苯甲酸)(干燥后约40 g)。滤液用几份饱和碳酸氢钠(NaHCO3)萃取(酸化后,该水洗液产生额外的间氯苯甲酸),并真空除去有机溶剂。将结晶残留物(重32 g,深色)溶于150 mL沸腾的甲醇中,加入18 g固体氢氧化钠,并在蒸汽浴上加热几分钟。将混合物加入800 mL水中,并用滤纸机械除去少量表面浮渣。溶液用浓盐酸酸化,析出30.9 g棕褐色固体。用水重结晶得到2,3-二甲基-4-甲氧基苯酚,为白色针状晶体,熔点为95-96 °C。分析元素(C9H12O2)H;C:计算值71.06;实测值70.20。N-甲基氨基甲酸酯是通过处理苯酚溶液(1 g溶于75 mL己烷并加入5 mL二氯甲烷)与2 g异氰酸甲酯和几滴三乙胺制成的。分离出的淡粉色固体用甲醇重结晶,得到熔点为141-142 °C的产物。分析元素(C11H15NO3):C,H,N。
向23.1 g片状氢氧化钾溶于250 mL热乙醇的溶液中,加入61.8 g 2,3-二甲基-4-甲氧基苯酚,随后加入60 g碘甲烷。将其回流12小时,然后真空除去溶剂。残留物溶于1.2 L水中,用盐酸酸化,并用3x200 mL二氯甲烷萃取。合并的萃取液用3x100 mL 5%氢氧化钠洗涤,真空除去溶剂。残留物在过滤和风干后形成重37.7 g的灰白色小叶状晶体块。用甲醇重结晶得到2,3-二甲基-1,4-二甲氧基苯,为白色固体,熔点为78-79 °C。分析元素(C10H14O2):C,H。探索了从2,3-二甲苯酚通过含氮中间体得到该二醚的另一条路线。该序列涉及2,3-二甲苯酚与亚硝酸反应(4-亚硝基产物,熔点184 °C分解),用连二亚硫酸钠还原(4-氨基产物,熔点约175 °C),用硝酸氧化(苯醌,熔点58 °C),用连二亚硫酸钠还原(对苯二酚),最后用碘甲烷甲基化。此过程的收率较差。
将88 g三氯氧磷和99 g N-甲基甲酰苯胺的混合物孵育至形成深紫红色,然后用36.5 g 2,3-二甲基-1,4-二甲氧基苯处理,并在蒸汽浴上加热3小时。然后将其倒入1 L水中,搅拌直至完全形成松散、易碎的深色结晶块。过滤除去,并溶于300 mL二氯甲烷中。先用水洗涤,再用5%氢氧化钠洗涤,最后用稀盐酸洗涤,真空除去溶剂,得到39.5 g凝固的黑色油状物。将其用2x300 mL沸腾的己烷萃取,合并萃取液,真空除去溶剂。淡黄色残留物结晶得到32.7 g 2,5-二甲氧基-3,4-二甲基苯甲醛,熔点为46-47 °C。反复用甲醇重结晶将熔点提高到59-60 °C。制备了丙二腈衍生物(醛和丙二腈在乙醇中加几滴三乙胺),为乙醇中析出的黄色晶体,熔点为190-191 °C。分析元素(C14H14N2O2)C,H;N:计算值11.56;实测值11.06, 11.04。
将16.3 g 2,5-二甲氧基-3,4-二甲基苯甲醛溶于50 mL硝基甲烷中,加入3.0 g无水乙酸铵,混合物在蒸汽浴上加热过夜。然后加入等体积的甲醇,冷却后得到一茬精细的黄色晶体。过滤除去,用甲醇洗涤,风干得到4.4 g 2,5-二甲氧基-3,4-二甲基-β-硝基苯乙烯,熔点为120-121 °C,用甲醇(50 mL/g)重结晶并未改善熔点。上述过滤的母液用水稀释至永久浑浊,然后置于冷藏箱中。沉积出块状、颗粒状、番茄红色的晶体,干燥后重2.5 g。其熔点为118-119.5 °C,与黄色样品的混合熔点未降低。两种形式具有相同的核磁共振谱(2.20, 2.25 CH3; 3.72, 3.84 OCH3; 6.80 ArH; 7.76, 8.28 CH=CH,具有14个循环分裂),红外光谱,紫外光谱(在乙醇中最大值324 nm,肩峰366 nm;在己烷中有两个峰309和355 nm)和微量分析。分析元素(C12H15NO4):C,H,N。
在氦气下,用外部冰浴将氢化铝锂(LAH)(56 mL 1 M四氢呋喃(THF)溶液)冷却至0 °C。在充分搅拌下,逐滴加入1.52 mL 100%硫酸(H2SO4),以尽量减少炭化。随后在1小时内加入3.63 g 2,5-二甲氧基-3,4-二甲基-β-硝基苯乙烯溶于36 mL无水四氢呋喃的溶液。再搅拌几分钟后,将温度升高至在蒸汽浴上温和回流约5分钟,然后再次冷却至0 °C。通过小心加入9 mL异丙醇(IPA),随后加入2.5 mL 15%氢氧化钠,最后加入7.5 mL水来破坏过量的氢化物。过滤反应混合物,滤饼先用四氢呋喃洗涤,然后用异丙醇洗涤。滤液真空除去溶剂,残留物在0.2 mm/Hg下于110-120 °C蒸馏,得到2.07 g 2,5-二甲氧基-3,4-二甲基苯乙胺,为清澈的白色油状物。将其溶于10 mL异丙醇中,用浓盐酸中和,然后用25 mL无水乙醚(Et2O)稀释。过滤形成的晶体,用乙醚洗涤,并风干至恒重。得到2.13 g美丽的白色2,5-二甲氧基-3,4-二甲基苯乙胺盐酸盐(2C-G)晶体,熔点为232-233 °C。分析元素(C12H20ClNO2):C,H。
给药剂量: 20 - 35 mg。
药效时长: 18 - 30 小时。
定性评论: (摄入22 mg)我完全能够正常运作,包括写作和接电话,但咖啡的味道真的很奇怪。虽然精神效应(仅达到++)级)正在消散,但身体对这一天仍有相当多的记忆。傍晚时分的睡眠很好,也很令人向往。
(摄入32 mg)极好的物质,应归类为“真正的致幻剂”,除非你是为了发表文章,在这种情况下,它最好被描述为“洞察力增强剂”,显然在心理治疗中具有潜在价值(如果有人愿意在治疗过程中花费30个小时!)。我想最好还是坚持其增强洞察力的作用,跳过心理治疗。实在是太长,太长了。对我来说,并没有特别的视觉冲击。非性欲和厌食方面可能会随着熟悉程度的增加而改变。拭目以待。当然,体验的时长不利于频繁使用,这很遗憾,因为这是一种非常值得尽可能经常研究的物质。
(摄入32 mg)就在一开始,上背部有一种非物理性的热感,这让我想起各种吲哚类药物起效时的感觉,但这并不是吲哚。能量颤动自始至终都很强烈,但身体不知何故总体上是放松的。
(摄入32 mg)两小时即达到平台期,肚子有点恶心。几个小时后我仍处于平台期。终于在第18个小时入睡,但即使第二天起床做了各种各样的事情,直到那天晚上我才完全恢复基线。如果要完全恢复,还需要几天时间。对于少量的物质来说,这是很长的里程。
延伸和评论: 这是有史以来第一个与其相关的三碳苯丙胺效力大致相同的苯乙胺例子。初步看来,从记录的笔记中很难区分2C-G和GANESHA本身在效力、持续时间或活性性质方面的任何主要差异。
我一直认为苯乙胺比相应的苯丙胺稍微弱一些。有时弱一点,有时弱很多。但这完全是一种带有偏见的观点,源于我最早对麦司卡林和TMA的比较。这种事情会影响一个人的思维,掩盖可能有价值的观察结果。同样有理由认为苯乙胺是原型,而苯丙胺比相应的苯乙胺稍微强一些。有时强一点,有时强很多。那么问题突然从问苯乙胺有什么不同,转变为问苯丙胺有什么不同?这只是一个历史事实,在我大部分的探索中,苯丙胺首先被制造和评估,因此倾向于扮演原型的角色。无论如何,在这里两者的效力趋于一致。