80 F-22¶
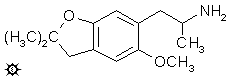
6-(2-AMINOPROPYL)-2,2-DIMETHYL-5-METHOXY-2,3-DIHYDROBENZOFURAN (6-(2-氨基丙基)-2,2-二甲基-5-甲氧基-2,3-二氢苯并呋喃)
 |
|---|
合成: 向 43.2 g 片状氢氧化钾溶于 250 mL 热乙醇的溶液中加入 96 g 4-甲氧基苯酚,随后在 2 小时内加入 90 g 2-甲基烯丙基氯。混合物回流 24 小时,然后加入 1.6 L 水中。加入足量的 25% 氢氧化钠使该相呈强碱性,然后用 3x200 mL 二氯甲烷提取。合并的提取液用水洗涤,并在真空下除去溶剂。残留物为 125 g 淡琥珀色油状物,即粗制 4-(2-甲基烯丙氧基)苯甲醚,无需进一步纯化即可用于后续反应。
在一个装有内部温度计的圆底烧瓶中,放置 125 g 未纯化的 4-(2-甲基烯丙氧基)苯甲醚,并用明火加热。当内部温度达到 190 °C 时,发生放热反应,使温度升至 250 °C,并在此保持 2 分钟。反应混合物冷却至室温后,倒入 500 mL 水中,用 25% 氢氧化钠强碱化,并用 100 mL 二氯甲烷反复提取,直至提取液基本无色。合并这些提取液并除去溶剂,得到 80.0 g 深色油状物,经证明主要是相应取代的二氢苯并呋喃。上述水相残留物用浓盐酸酸化,再次用二氯甲烷提取。除去溶剂得到 17.7 g 4-甲氧基-2-(2-甲基烯丙基)苯酚,为琥珀色油状物,最终凝固为白色晶体,熔点为 52.5-54 °C。
将 17 g 4-甲氧基-2-(2-甲基烯丙基)苯酚溶于 56 g 乙酸的溶液中,用 8.4 g 氯化锌处理,随后加入 28 mL 浓盐酸。该混合物用加热套在回流温度下加热 1 小时。冷却后,将其倒入水中并用 2x150 mL 二氯甲烷提取。合并的提取液用几份 8% 氢氧化钠洗涤,直至提取液无色。然后用水洗涤有机部分,除去溶剂得到 5.8 g 2,2-二甲基-5-甲氧基-2,3-二氢苯并呋喃,为具有刺激性气味的淡琥珀色油状物。通过蒸馏纯化,得到沸点为 136-138 °C (33 mm/Hg) 的灰白色油状馏分。
向静置 0.5 小时的 8.0 g N-甲基甲酰苯胺和 9.2 g 三氯氧磷的混合物中,加入 4.0 g 2,2-二甲基-5-甲氧基-2,3-二氢苯并呋喃,混合物在蒸汽浴温度下保持 2.5 小时。然后将其倒入 200 mL 水中,产生黑色油相,没有任何结晶迹象。该混合物用 3x150 mL 二氯甲烷提取,合并的提取液在真空下除去溶剂。残留的油状物(经气相色谱显示含有大约等量的两种苯甲醛异构体 A 和 B)用三份 75 mL 沸腾己烷提取,每份冷却后都会析出一种部分结晶的微红色油状物。第四次己烷提取没有得到更多东西。从这三种提取物中倾析出溶剂,半固体残留物在 3.0 mL 甲醇中研磨,得到 1.4 g 2,2-二甲基-6-甲酰基-5-甲氧基-2,3-二氢苯并呋喃(异构体 RBS)的淡黄色晶体。从甲醇重结晶后,颜色几乎为白色,熔点为 79.5-80.5 °C。合并的母液富含异构体 RAS,经制备型气相色谱分离和核磁共振分析证明是 7-甲酰基异构体。从上述克莱森重排中分离出的 80 g 不纯二氢苯并呋喃经蒸馏,沸点为 138-153 °C (30 mm/Hg) 的馏分 (43.8 g) 按此所述处理成醛混合物。经过类似的己烷提取,最终获得了 4.0 g 纯度为 95% 的异构体 RBS。该馏分的其余成分尚未确定,但可能含有一些具有六元苯并吡喃环系统的成分。
向 5.2 g 2,2-二甲基-6-甲酰基-5-甲氧基-2,3-二氢苯并呋喃溶于 20 mL 冰醋酸的溶液中,加入 3 mL 硝基乙烷,随后加入 1.6 g 无水醋酸铵。该混合物在蒸汽浴上加热 4 小时,然后向热溶液中加入少量水。这促使大量砖红色晶体沉积,冷却后过滤除去,并从 50 mL 沸腾甲醇中重结晶。风干后,由此得到 2.7 g 荧光般的美味橙色晶体,即 2,2-二甲基-5-甲氧基-6-(2-硝基-1-丙烯基)-2,3-二氢苯并呋喃。通过处理母液又获得了 0.6 g 产物。
在 300 mL 回流的无水乙醚中悬浮 2.5 g 氢化铝锂,用 3.1 g 2,2-二甲基-5-甲氧基-6-(2-硝基-1-丙烯基)-2,3-二氢苯并呋喃的乙醚溶液处理。混合物在回流温度下保持 18 小时。冷却后,小心加入含有 15 g 硫酸的 400 mL 水破坏过量的氢化物。分离水相,用乙醚洗涤一次,然后用二氯甲烷洗涤一次。然后加入 60 g 酒石酸钾钠,并通过加入 25% 氢氧化钠将 pH 调至 10 以上。用 3x250 mL 二氯甲烷提取,合并提取液,并在真空下除去溶剂。残留 2.8 g 具有氨味的琥珀色油状物。将其溶解在 200 mL 无水乙醚中,并通入无水氯化氢气体至饱和。立即形成油状物,从中倾析出上清液乙醚。残留的油状物重新悬浮在第二份 200 mL 无水乙醚中,再次倾析,最后第三份 200 mL 乙醚使剩余的油状物溶解,得到澄清溶液。所有三种溶液在接下来的几个小时内都变成了胶状,并且每一种在接下来的几天内都析出了一批白色晶体。从第一份中得到 1.4 g 产物,熔点为 153-154 °C;从第二份中得到 0.2 g,熔点为 153-154 °C;从第三份中得到 1.2 g,熔点为 155-156 °C。合并这些晶体,并从 10 mL 沸腾乙腈中重结晶,得到 1.7 g 6-(2-氨基丙基)-2,2-二甲基-5-甲氧基-2,3-二氢苯并呋喃盐酸盐 (F-22),为白色结晶固体,熔点为 154-155 °C。这种材料即使在干燥时,也表现出随时间变色的倾向。
给药剂量: 大于 15 mg。
药效时长: 未知。
延伸和评论: 这是又一种二氢苯并呋喃,如果它真的有活性的话,其效力也不是很高。这种特殊的二氢苯并呋喃类似物 F-22 曾让我觉得特别有可能成为活性候选者。它有一种特殊的韵律。F-22,就像 LSD-25。它就在我必须向大麻大会提交关于所有这些二氢苯并呋喃的合成(和活性!)的论文前五天完成了。这可能是另一种 LSD 吗?我完全相信这种可能性是真实存在的,所以我实际上以 10 微克的异常低水平开始了筛选过程。两天后,我将剂量提高到 25 微克(再次没有活性),三天后,在飞往哥本哈根的极地航班上,凌晨 1 点,我吞下了 50 微克的“巨量”剂量。孤注一掷。如果我在 SAS 飞机的旅游区全面发作,那将是一篇相当精彩的论文。如果没有,我总是可以说类似“尚未发现活性水平”之类的话。没有活性。另一个白日梦般的幻想破灭了。
事实证明,整个项目基本上已经耗尽了动力。许多巧妙的类似物已经开始制作,如果这些二氢苯并呋喃中有任何一种显示出任何形式的活性,它们就会被继续研究。上述“另一种”苯甲醛可以用类似于 F-2 对应物所提出的方式进行处理,以制备最终的苯丙胺,即 7-(2-氨基丙基)-2,2-二甲基-5-甲氧基-2,3-二氢苯并呋喃。在通往 F-233 的道路上取得了长足的进步(我已经在 F-2 下讨论了命名系统,F 代表苯并呋喃的呋喃,2、3 和 3 代表其上甲基的位置)。4-甲氧基苯酚与 1-氯-3-甲基-2-丁烯反应生成醚,该醚经热克莱森重排生成 2-(1,1-二甲基烯丙基)-4-甲氧基苯酚,沸点为 148-157 °C (30 mm/Hg)。将其环化为中间体环 2,3,3-三甲基-2,3-二氢苯并呋喃,蒸馏后经气相色谱分析显示纯度仅为 80%。尽管如此(怀着年轻无辜化学家骨子里的希望),还是将其推进到苯甲醛阶段(在产生的拒绝结晶的油状物中发现了四种苯甲醛,这并不太令人惊讶)。然后(当绝望取代了希望时),这些苯甲醛与硝基乙烷缩合形成更糟糕的混合物。也许能从中结晶出什么东西?什么也没有。垃圾。所有的东西都被简单地放在架子上,至今仍在那里,而 F-233,即 6-(2-氨基丙基)-5-甲氧基-2,3,3-三甲基-2,3-二氢苯并呋喃,仍然是推测的东西。
通往 F-23(6-(2-氨基丙基)-2,3-二甲基-5-甲氧基-2,3-二氢苯并呋喃)的道路也只走到了起始醚这一步,这时我突然想到最终产物将具有前所未有的三个手性中心,因此总共有四对消旋非对映异构体。然后我发现起始烯丙基卤化物,巴豆基氯,纯度只有 80%,其余 20% 是 3-氯-1-丁烯。这最终会产生 2-乙基类似物,即 6-(2-氨基丙基)-2-乙基-5-甲氧基-2,3-二氢苯并呋喃,它有两个手性中心和另外两对立体异构体(更不用说需要设计一套全新的编码系统了)。除非有什么结晶中间体掉进我的腿上,否则最终的烂摊子里面至少会有六种离散的化合物,甚至还没考虑光学异构体。我甚至还没开始考虑制造六元二氢苯并吡喃,也就是最初提出整个项目理由的 THC 类似物。最近一期的《药物化学杂志》刚刚发表了一篇文章,描述了 6-甲氧基四氢苯并吡喃与二氯甲基甲醚的反应,获得了大约等量的所有三种可能的异构体。那将是制造原型化合物 7-(2-氨基丙基)6-甲氧基-1,2,3,4-四氢苯并吡喃的第一步。就像苯并呋喃都被命名为 F 化合物一样,作为苯并吡喃,这本来应该是 P 化合物,但 P 也用于普罗斯卡林 (proscaline),这些代码需要进行一些修复工作。
是时候弃船了。大约在这个时候,我刚刚合成并发现了 ARIADNE 的奇怪活性,这使得弃船变得容易接受得多。